
RECARBRIO 500 mg-500 mg-250 mg, poudre pour solution pour perfusion, boîte de 25 flacons de 100 ml
Dernière révision : 11/10/2021
Taux de TVA : 2.1%
Laboratoire exploitant : MSD FRANCE
Source :

Recarbrio est indiqué dans :
Le traitement des pneumonies nosocomiales (PN), dont les pneumonies acquises sous ventilation mécanique (PAVM) chez les adultes (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Le traitement d'une bactériémie associée, ou suspectée d'être associée à une PN ou une PAVM chez les adultes.
Le traitement des infections dues à des bactéries aérobies à Gram négatif chez les adultes pour qui les options thérapeutiques sont limitées (voir rubriques Posologie et mode d'administration, Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
Hypersensibilité aux substances actives ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Hypersensibilité à tout autre antibiotique de la classe des carbapénèmes.
Hypersensibilité sévère (par exemple, réaction anaphylactique, réaction cutanée sévère) à tout autre antibiotique de la famille des bêta-lactamines (par exemple, pénicillines, céphalosporines ou monobactames) (voir rubrique Mises en garde spéciales et précautions d'emploi).
Réactions d'hypersensibilité
Des réactions d'hypersensibilité (anaphylactiques) graves et parfois fatales ont été rapportées chez des patients traités par des bêta-lactamines (voir rubriques Contre-indications et Effets indésirables).
Ces réactions surviennent plus fréquemment chez des patients ayant des antécédents de réactions d'hypersensibilité à plusieurs allergènes. Avant d'instaurer un traitement par Recarbrio, un interrogatoire attentif doit rechercher des antécédents de réaction d'hypersensibilité aux carbapénèmes, aux pénicillines, aux céphalosporines, à d'autres bêta-lactamines et à d'autres allergènes.
La survenue d'une réaction allergique à Recarbrio impose l'arrêt immédiat du traitement par Recarbrio. Les réactions anaphylactiques graves nécessitent l'instauration immédiate d'un traitement d'urgence.
Fonction hépatique
La fonction hépatique doit être étroitement surveillée pendant le traitement par Recarbrio en raison du risque de toxicité hépatique (tel qu'une augmentation des transaminases, une insuffisance hépatique et une hépatite fulminante) (voir rubrique Effets indésirables).
Utilisation chez les patients présentant une maladie hépatique : les patients ayant des troubles hépatiques préexistants doivent faire l'objet d'une surveillance de la fonction hépatique pendant le traitement par Recarbrio. Aucune adaptation posologique n'est requise (voir rubrique Posologie et mode d'administration).
Système nerveux central (SNC)
Des effets indésirables sur le SNC, tels que des convulsions, des états confusionnels et une activité myoclonique ont été rapportés lors du traitement par imipénème/cilastatine, des composants de Recarbrio, en particulier en cas d'administration d'imipénème à des doses supérieures aux doses recommandées. Ces cas ont été rapportés plus fréquemment chez les patients présentant des troubles du SNC (par exemple lésions cérébrales ou antécédents de convulsions) et/ou une altération de la fonction rénale.
Augmentation du risque de convulsion liée à l'interaction avec l'acide valproïque
L'administration concomitante de Recarbrio et d'acide valproïque/valproate de sodium est déconseillée. Des antibiotiques n'appartenant pas à la famille des carbapénèmes doivent être envisagés pour le traitement des infections chez les patients dont les crises convulsives sont bien contrôlées sous acide valproïque ou valproate de sodium. Si l'administration de Recarbrio est nécessaire, un traitement anticonvulsivant supplémentaire doit être envisagé (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Diarrhée associée à Clostridioides difficile (DACD)
Une diarrhée associée à Clostridioides difficile (DACD) a été rapportée avec Recarbrio. La sévérité d'une DACD peut varier d'une forme légère à la mise en jeu du pronostic vital. Ce diagnostic de DACD doit être envisagé chez tous les patients présentant une diarrhée pendant ou après l'administration de Recarbrio (voir rubrique Effets indésirables). Un examen attentif des antécédents médicaux est nécessaire dans la mesure où il a été rapporté une survenue de DACD plus de deux mois après l'administration d'antibiotiques.
En cas de DACD suspectée ou confirmée, l'arrêt du traitement par Recarbrio et l'administration d'un traitement spécifique vis-à-vis de C. difficile doivent être envisagés. Les médicaments inhibant le péristaltisme ne doivent pas être administrés.
Patients présentant une ClCr ≥ 150 mL/min
Sur
la base de l’analyse de la relation
pharmacocinétique/pharmacodynamique, la dose de Recarbrio recommandée
chez les patients présentant une ClCr ≥ 90 mL/min peut ne pas être
suffisante pour traiter les patients présentant une PN ou une PAVM et
une ClCr > 250 mL/min., ou les patients présentant une IIAc ou IUc
et une ClCr > 150 mL/min. Il conviendrait d’envisager l’utilisation
d’alternatives thérapeutiques pour ces patients.
Insuffisance rénale
Une adaptation posologique est recommandée chez les patients présentant
une insuffisance rénale (voir rubrique Posologie et mode
d'administration). Les données disponibles sont insuffisantes pour
recommander l’utilisation de Recarbrio chez les patients sous dialyse
péritonéale.
Limites des données cliniques
Les patients immunodéprimés, y compris ceux présentant une neutropénie, ont été exclus des essais cliniques.
Pneumonies nosocomiales, dont les pneumonies acquises sous ventilation mécanique
Dans une étude unique menée dans les pneumonies nosocomiales, dont les
pneumonies acquises sous ventilation mécanique, 6,2 % (33/535) des
patients présentaient une bactériémie à l’inclusion.
Patients pour qui les options thérapeutiques sont limitées
L’utilisation de Recarbrio pour traiter des patients présentant des
infections dues à des bactéries aérobies à Gram négatif pour qui les
options thérapeutiques sont limitées est basée sur l’expérience avec
l’imipénème/cilastatine, l’analyse de la relation
pharmacocinétique/pharmacodynamique de
l’imipénème/cilastatine/relebactam, et sur des données limitées issues
d’une étude clinique randomisée dans laquelle 21 patients évaluables
ont été traités par Recarbrio et 10 patients évaluables ont été traités
par la colistine et l’imipénème/cilastatine pour des infections causées
par des bactéries non
sensibles à l’imipénème.
Limites du spectre d'activité antibactérienne
L'imipénème n'exerce aucune activité contre Staphylococcus aureus résistant à la méticilline (SARM) et Staphylococcus epidermidis résistant à la méticilline (SERM) ou contre Enterococcus faecium. Des antibiotiques différents ou additionnels doivent être utilisés si ces pathogènes sont connus ou suspectés d'être impliqués dans l'infection.
Le spectre d'inhibition du relebactam inclut les bêta-lactamases de classe A (notamment BLSE et KPC) et les bêta-lactamases de classe C dont PDC. Le relebactam n'inhibe pas les carbapénémases de classe D telles qu'OXA-48 ou les métallo-bêta-lactamases de classe B telles que NDM et VIM (voir rubrique Propriétés pharmacodynamiques).
Micro-organismes non sensibles
L'utilisation d'imipénème/cilastatine/relebactam peut favoriser la prolifération de micro-organismes non sensibles, ce qui peut nécessiter l'interruption du traitement ou d'autres mesures appropriées.
Séroconversion du test à l'antiglobuline (test de Coombs)
Un test de Coombs direct ou indirect peut devenir positif au cours du traitement par l'imipénème/cilastatine/relebactam (voir rubrique Effets indésirables).
Régime contrôlé en sodium
Chaque flacon contient au total 37,5 mg de sodium (1,6 mmol), ce qui équivaut à 1,9 % de l'apport alimentaire quotidien maximal recommandé par l'OMS (Organisation Mondiale de la Santé) de 2 g de sodium par adulte. Ceci doit être pris en compte lors de l'administration de Recarbrio à des patients qui suivent un régime hyposodé.
Résumé du profil de sécurité
L'effet indésirable le plus fréquent (≥ 2 %) survenu chez les patients recevant l'imipénème/cilastatine
plus relebactam dans les essais de phase II groupés menés dans les
infections intra-abdominales compliquées (IIAc) et les infections
urinaires compliquées (IUc), dont les pyélonéphrites (N = 431) a été la
diarrhée. Les effets indésirables les plus fréquents (≥ 2
%) survenus chez les patients recevant Recarbrio dans un essai de phase
III mené dans la PN ou la PAVM (N = 266) ont été la diarrhée, l'augmentation de l'alanine aminotransférase et l'augmentation de l'aspartate aminotransférase.
Résumé des effets indésirables
Les effets indésirables suivants ont été rapportés au cours des essais cliniques de phase II (imipénème/cilastatine plus relebactam incluant 431 patients) et de phase III (Recarbrio incluant 266 patients) dans le cadre des études cliniques avec imipénème/cilastatine ou depuis la commercialisation de l'imipénème/cilastatine (voir Tableau 3).
Les effets indésirables sont présentés par classes de systèmes d'organes MedDRA et par fréquence. Les catégories de fréquence sont définies selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000), et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 3 : Fréquence des effets indésirables par classes de systèmes d'organes
| Classes de systèmes d'organes | Fréquent | Peu fréquent | Rare | Très rare | Fréquence indéterminée |
| Infections et infestations | | | Colite pseudomembraneu se* | Gastro-entérite* | |
| | Candidose* | | |||
| | | Pancytopénie* | | | |
| Affections hématologiques et du système lymphatique | Éosinophilie* | Neutropénie* Leucopénie* Thrombocytopéni e* | Agranulocytose* | Anémie hémolytique* Aplasie médullaire* | |
| | | Thrombocytose* | | | |
| Affections du système immunitaire | | | Réactions anaphylactiques* | | |
| | | Convulsions* | | | |
| | Hallucinations* | Encéphalopathie* | | | |
| Affections du système nerveux | Etats confusionnels* Activité myoclonique* | Paresthésies* Tremblement focal* | Aggravation d'une myasthénie grave* Céphalées* | Agitation* Dyskinésie* | |
| | Etourdissements* | Dysgueusie* | | | |
| | Somnolence* | | | |
| Classes de systèmes d'organes | Fréquent | Peu fréquent | Rare | Très rare | Fréquence indéterminée |
| Affections de l'oreille et du labyrinthe | | | Perte d'audition* | Vertiges* Acouphènes* | |
| Affections cardiaques | | | | Cyanose* Tachycardie* Palpitations* | |
| Affections vasculaires | Thrombophlébite* | Hypotension* | | Bouf ées vasomotrices* | |
| Affections respiratoires, thoraciques et médiastinales | | | | Dyspnée* Hyperventilation* Douleur pharyngée* | |
| Affections gastro- intestinales | Diarrhée†* Nausées†* Vomissements†* | | Coloration des dents et/ou de la langue* | Colite hémorragique* Douleurs abdominales* Pyrosis* Glossite* Hypertrophie des papilles linguales* Hypersalivation* | |
| Affections hépatobiliaires | Elévation de l'alanine aminotransférase†* Elévation de l'aspartate aminotransférase†* | | Insuffisance hépatique* Hépatite* | Hépatite fulminante * | Ictère* |
| Affections de la peau et du tissu sous-cutané | Eruption cutanée (par exemple, exanthématheux)* | Urticaire* Prurit* | Nécrolyse épidermique toxique * Angio-œdème * Syndrome de Stevens-Johnson * Erythème multiforme* Dermatite exfoliative* | Hyperhidrose* Modifications de la texture de la peau * | |
| Affections musculo- squelettiques et systémiques | | | | Polyarthralgies* Dorsalgies* | |
| Classes de systèmes d'organes | Fréquent | Peu fréquent | Rare | Très rare | Fréquence indéterminée |
| Affections du rein et des voies urinaires | | Augmentations du taux de créatinine sérique* | Insuffisance rénale aigüe* Oligurie/anurie* Polyurie* Coloration anormale des urines (anodine et ne devant pas être confondue avec une hématurie)* | | |
| Affections des organes de reproduction et du sein | | | | Prurit vulvaire* | |
| Troubles généraux et anomalies au site d'administration | | Fièvre* Douleur locale et induration au site d'injection* | | Gêne thoracique* Asthénie/ faiblesse* | |
| Investigations | Elévations des phosphatases alcalines sériques* | Test de Coombs positif* Allongement du temps de prothrombine* Diminution de l'hémoglobine* Elévation de la bilirubinémie* Elévation de l'urée sanguine* | | | Elévation du taux sanguin de lactate déshydrogéna se* |
| *Effets rapportés avec l'imipénème/cilastatine dans le cadre des études cliniques ou depuis la commercialisation de l'imipénème/cilastatine †Effets rapportés avec l'imipénème/cilastatine plus relebactam au cours des études de phase II (N = 431) et de phase III (N = 266) | |||||
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT l'instauration du traitement :
- Rechercher les antécédents de réaction d'hypersensibilité aux carbapénèmes, aux pénicillines, aux céphalosporines, à d'autres bêta-lactamines et à d'autres allergènes.
SURVEILLANCE pendant le traitement :
- Fonction hépatique.
Un test de Coombs direct ou indirect peut devenir positif au cours du traitement par l'imipénème/cilastatine/relebactam.
- Rechercher les antécédents de réaction d'hypersensibilité aux carbapénèmes, aux pénicillines, aux céphalosporines, à d'autres bêta-lactamines et à d'autres allergènes.
SURVEILLANCE pendant le traitement :
- Fonction hépatique.
Un test de Coombs direct ou indirect peut devenir positif au cours du traitement par l'imipénème/cilastatine/relebactam.
Grossesse
Il n'a pas été mené d'études appropriées et bien contrôlées concernant l'utilisation d'imipénème, de cilastatine ou de relebactam chez la femme enceinte.
Les études menées chez l'animal avec l'imipénème/cilastatine ont montré une toxicité sur la reproduction chez les singes (voir rubrique Données de sécurité préclinique). Le risque potentiel chez l'Homme n'est pas connu. Les études menées chez l'animal avec le relebactam n'ont pas mis en évidence d'effets délétères directs ou indirects au regard de la toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Recarbrio ne doit être utilisé pendant la grossesse que si le bénéfice potentiel l'emporte sur le risque potentiel pour le fœtus.
Allaitement
L'imipénème et la cilastatine sont excrétés en faibles quantités dans le lait maternel.
On ne sait pas si le relebactam est excrété dans le lait maternel. Des données disponibles chez l'animal ont montré que le relebactam est excrété dans le lait de rates (voir rubrique Données de sécurité préclinique pour plus de détails).
Un risque pour les nouveau-nés/nourrissons allaités ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement avec Recarbrio en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Aucune donnée n'est disponible chez l'Homme concernant les ef ets potentiels de imipénème/cilastatine ou relebactam sur la fertilité masculine ou féminine. Les études menées chez l'animal n'ont pas mis en évidence d'effets délétères de l'imipénème/cilastatine ou du relebactam sur la fertilité (voir rubrique Données de sécurité préclinique).
Ganciclovir
Des convulsions généralisées ont été rapportées chez des patients qui recevaient de façon concomitante du ganciclovir et de l'imipénème/cilastatine, des composants de Recarbrio. Le ganciclovir ne doit être administré de façon concomitante avec Recarbrio que si le bénéfice potentiel est supérieur aux risques.
Acide valproïque
Des cas rapportés dans la littérature ont montré que, chez des patients recevant de l'acide valproïque ou du valproate de sodium, l'administration concomitante de carbapénèmes, y compris d'imipénème/cilastatine (des composants de Recarbrio), entraîne une diminution des concentrations d'acide valproïque. Les concentrations d'acide valproïque peuvent diminuer à des valeurs inférieures à l'intervalle thérapeutique du fait de cette interaction, ce qui augmente par conséquent le risque de crises convulsives. Bien que le mécanisme de cette interaction soit inconnu, les données d'études menées in vitro et chez l'animal suggèrent que les carbapénèmes peuvent inhiber l'hydrolyse du métabolite glucuronide de l'acide valproïque (VPA-g) pour reformer l'acide valproïque, diminuant ainsi les concentrations sériques d'acide valproïque. L'administration concomitante de Recarbrio et d'acide valproïque/valproate de sodium est déconseillée (voir rubrique Mises en garde spéciales et Précautions d'emploi).
Anticoagulants oraux
L'administration simultanée d'antibiotiques avec la warfarine peut augmenter les effets anticoagulants de cette dernière. Il est recommandé de contrôler l'INR de façon appropriée pendant et juste après l'administration concomitante d'antibiotiques et d'anticoagulants oraux.
Études cliniques d'interaction médicamenteuse
Une étude clinique d'interaction médicamenteuse a montré que l'exposition à l'imipénème et au relebactam n'augmente pas de façon cliniquement significative lorsque Recarbrio est administré de façon concomitante avec le probénécide, inhibiteur d'OAT, ce qui indique l'absence d'interactions médicamenteuses cliniquement significatives médiées par les OAT. L'administration concomitante d'imipénème/cilastatine et de probénécide a augmenté la concentration plasmatique et la demi-vie de la cilastatine, toutefois pas de façon cliniquement significative. Par conséquent, Recarbrio peut être administré de façon concomitante avec des inhibiteurs d'OAT.
Il est recommandé que Recarbrio soit utilisé pour traiter des infections dues à des bactéries aérobies à Gram négatif chez des patients adultes pour qui les options thérapeutiques sont limitées, uniquement après avis d'un médecin expérimenté dans la prise en charge des maladies infectieuses.
Posologie
Le Tableau 1 indique la posologie recommandée pour l'administration intraveineuse chez les patients présentant une clairance de la créatinine (ClCr) ≥ 90 mL/min (voir rubriques Mises en garde spéciales et Précautions d'emploi et Propriétés pharmacodynamiques).
Tableau 1 : Posologie recommandée pour l'administration intraveineuse chez les patients présentant une clairance de la créatinine (ClCr) ≥ 90 mL/min1,2
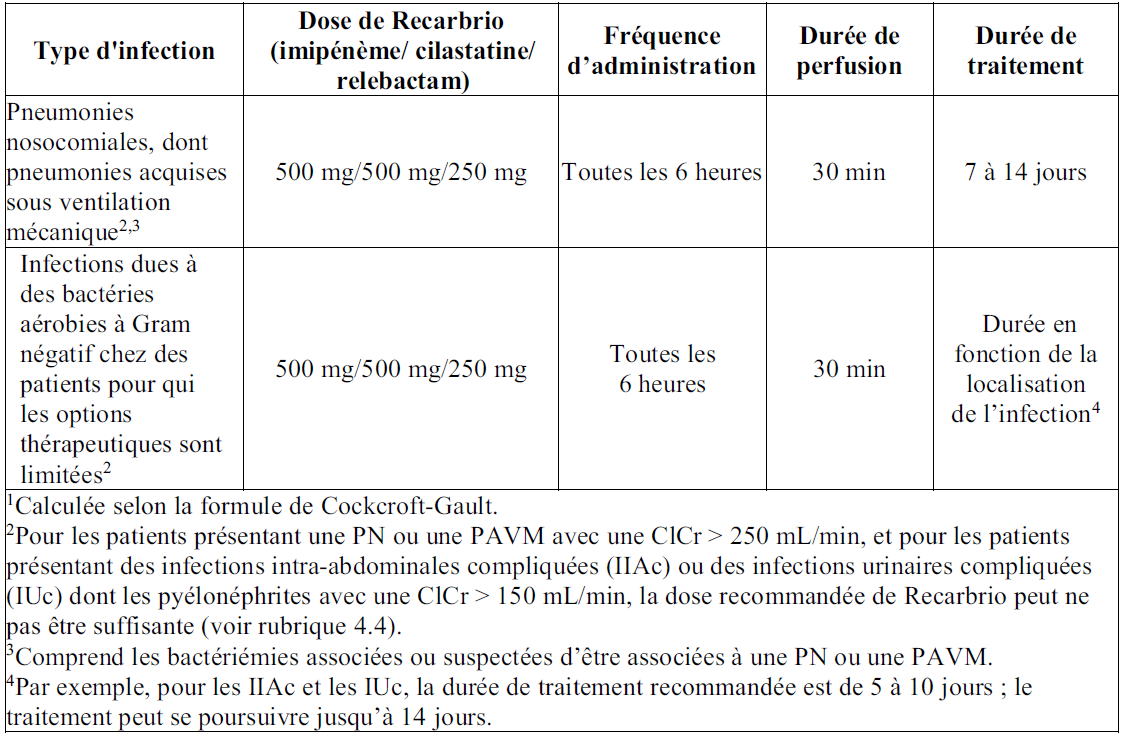
Populations particulières
Insuffisance rénale
Chez les patients présentant une ClCr inférieure à 90 mL/min, une réduction de la posologie de Recarbrio est nécessaire, comme indiqué dans le Tableau 2. Pour les patients ayant une fonction rénale variable, la ClCr doit être surveillée.
Tableau 2 : Posologies recommandées pour l'administration intraveineuse chez les patients présentant une ClCr < 90 mL/min
|
Clairance estimée de la créatinine (mL/min)* |
Posologie recommandée de Recarbrio (imipénème/cilastatine/relebactam) (mg)† |
|
Inférieure à 90 et supérieure ou égale à 60 |
400/400/200 |
|
Inférieure à 60 et supérieure ou égale à 30 |
300/300/150 |
|
Inférieure à 30 et supérieure ou égale à 15 |
200/200/100 |
|
Insuffisance rénale terminale (IRT) sous hémodialyse‡ |
200/200/100 |
|
*ClCr calculée selon la formule de Cockroft-Gault. †Administration par voie intraveineuse en 30 minutes toutes les 6 heures. ‡L'administration doit être prévue à la suite de l'hémodialyse. L'imipénème, la cilastatine et le relebactam sont éliminés de la circulation pendant l'hémodialyse. Recarbrio est fourni sous forme d'association à dose fixe dans un flacon unique ; la dose de chaque composant sera ajustée dans les mêmes proportions lors de la préparation (voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination). |
|
Recarbrio ne doit être administré chez les patients présentant une ClCr inférieure à 15 mL/min que si une hémodialyse est instaurée dans les 48 heures. Les données disponibles sont insuffisantes pour recommander l'utilisation de Recarbrio chez les patients sous dialyse péritonéale.
Insuffisance hépatique
Aucune adaptation posologique n'est requise chez les patients présentant une insuffisance hépatique (voir rubrique Propriétés pharmacocinétiques).
Population âgée
Aucune adaptation posologique n'est requise chez les patients âgés (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de l'imipénème/cilastatine/relebactam n'ont pas encore été établies chez les enfants et adolescents âgés de moins de 18 ans. Aucune donnée n'est disponible.
Mode d'administration
Voie intraveineuse.
Recarbrio est administré par perfusion intraveineuse de 30 minutes.
Recarbrio doit être reconstitué (voir rubriques Incompatibilités, Durée de conservation et Instructions pour l'utilisation, la manipulation et l'élimination) avant la perfusion intraveineuse.
Durée de conservation :
Poudre sèche
30 mois.
Après reconstitution et dilution
Les solutions diluées doivent être utilisées immédiatement. L'intervalle de temps entre le début de la reconstitution et la fin de la perfusion intraveineuse ne doit pas dépasser 2 heures.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation liées à la température. Conserver les flacons dans l'emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution et dilution, voir rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d'élimination et de manipulation.
En cas de surdosage, arrêter Recarbrio, traiter les symptômes et initier un traitement d'appoint.
L'imipénème, la cilastatine et le relebactam peuvent être éliminés par hémodialyse. Aucune information clinique n'est disponible sur l'utilisation de l'hémodialyse pour traiter un surdosage.
Classe pharmacothérapeutique : Antibactériens à usage systémique, carbapénèmes, code ATC : J01DH56
Mécanisme d'action
L'imipénème exerce une activité bactéricide en inhibant la synthèse du peptidoglycane de la paroi cellulaire bactérienne par fixation aux protéines de liaison aux pénicillines (PLP).
La cilastatine limite le métabolisme rénal de l'imipénème et ne possède pas d'activité antibactérienne.
Le relebactam est un inhibiteur non bêta-lactame des bêta-lactamases de classes A et C de Ambler, dont les carbapénémases Klebsiella pneumoniae carbapénémase (KPC) et les bêta-lactamases à spectre étendu (BLSEs) de classe A, et les bêta-lactamases de classe C (de type AmpC) dont les céphalosporinases produites par Pseudomonas (PDC). Le relebactam n'inhibe pas les enzymes de classe B (métallo-bêta-lactamases) ou les carbapénémases de classe D. Le relebactam n'a pas d'activité antibactérienne.
Résistance
Les mécanismes de résistance chez les bactéries à Gram négatif connus pour affecter l'imipénème/relebactam comprennent la production de métallo-bêta-lactamases ou d'oxacillinases avec une activité de carbapénémase.
L'expression de certains allèles de la bêta-lactamase à spectre étendu GES (Guiana extended- spectrum-bêta-lactamase) de classe A ou la surexpression de PDC couplées à une perte de porine OprD, voie d'entrée de l'imipénème, peuvent conférer une résistance à l'imipénème/relebactam chez P. aeruginosa. L'expression de pompes à efflux chez P. aeruginosa n'a d'incidence ni sur l'activité de l'imipénème, ni sur celle du relebactam. Les mécanismes de résistance bactérienne des Enterobacterales susceptibles de diminuer l'activité antibactérienne de l'imipénème/relebactam incluent des mutations de porines affectant la perméabilité de la membrane externe.
Activité antibactérienne en association avec d'autres antibiotiques
Des études in vitro n'ont
pas montré d'antagonisme entre l'imipénème/relebactam et l'amikacine,
l'azithromycine, l'aztréonam, la colistine, la gentamicine, la
lévofloxacine, le linézolide, la tigécycline, la tobramycine, ou la vancomycine.
Concentrations critiques
Les valeurs critiques des concentrations minimales inhibitrices (CMI) établies par l'European Committee on Antimicrobial Susceptibility Testing (EUCAST) sont les suivantes :
| Groupe d'organismes | Concentrations Minimales Inhibitrices (mg/L) | |
| Sensibles ≤ | Résistants > | |
| Enterobacterales (sauf Morganellaceae) | 2 | 2 |
| Pseudomonas aeruginosa | 2 | 2 |
| Acinetobacter spp. | 2 | 2 |
| Streptocoques du groupe viridans | 2 | 2 |
| Bactéries anaérobies à Gram positif | 2 | 2 |
| Bactéries anaérobies à Gram négatif | 2 | 2 |
Relation pharmacocinétique/pharmacodynamique
Il a été démontré que le pourcentage de temps pendant lequel les concentrations plasmatiques d'imipénème sous forme libre se situent au-dessus de la concentration minimale inhibitrice de l'imipénème/relebactam (% fT > CMI) est le paramètre le mieux corrélé à l'efficacité. Il a été déterminé que le rapport de l'ASC du relebactam sous forme libre dans le plasma à 24 heures sur la CMI de l'imipénème/relebactam (fASC/CMI) est l'index qui prédit le mieux l'activité du relebactam.
Efficacité clinique vis-à-vis de bactéries pathogènes spécifiques
L'efficacité a été démontrée dans des études cliniques vis-à-vis de bactéries pathogènes listées selon l'indication, celles-ci étant sensibles in vitro à l'imipénème et au relebactam :
Pneumonies nosocomiales, dont les pneumonies acquises sous ventilation mécanique
Bactéries à Gram négatif
Escherichia coli
Haemophilus influenzae
Klebsiella pneumoniae
Pseudononas aeruginosa
Serratia marcescens
Les études in vitro suggèrent que les pathogènes suivants seraient sensibles à l'imipénème et au relebactam en l'absence de mécanismes de résistance acquis :
Bactéries aérobies à Gram négatif
complexe Acinetobacter baumannii-calcoaceticus
Citrobacter spp. (dont C. freundii et C. koseri)
Enterobacter spp. (dont E. asburiae et E. cloacae)
Escherichia coli
Klebsiella spp. (dont K. aerogenes, K. oxytoca et K. pneumoniae)
Pseudomonas aeruginosa
Serratia marcescens
Bactéries anaérobies à Gram négatif
Bacteroides spp. (dont B. fragilis)
Fusobacterium spp. (dont F. nucleatum et F. necrophorum)
Prevotella spp. (dont P. melaninogenica, P. bivia et P. buccae)
Bactéries aérobies à Gram positif
Enterococcus faecalis
Staphylococcus aureus (seulement les isolats sensibles à la méticilline)
Streptocoques du groupe viridans (dont S. anginosus et S. constellatus)
Des études in vitro indiquent que les espèces suivantes ne sont pas sensibles à l'imipénème et au relebactam :
Bactéries aérobies à Gram négatif
Legionella spp.
Stenotrophomonas maltophilia
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Recarbrio dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement des infections bactériennes dues à des bactéries à Gram négatif (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Introduction générale
Les paramètres pharmacocinétiques à l'état d'équilibre de l'imipénème, la cilastatine et le relebactam chez des adultes sains avec une fonction rénale normale (ClCr égale ou supérieure à 90 mL/min), après plusieurs perfusions intraveineuses de 30 minutes de 500 mg d'imipénème/500 mg de cilastatine + 250 mg de relebactam administrées toutes les 6 heures sont résumés dans le Tableau 4. Les paramètres pharmacocinétiques à l'état d'équilibre de l'imipénème et du relebactam chez des patients présentant une IIAc ou une IUc et une PN ou une PAVM avec une fonction rénale normale (90 mL/min ≤ ClCr < 150 mL/min) après plusieurs perfusions intraveineuses de 30 minutes de 500 mg d'imipénème/500 mg de cilastatine + 250 mg de relebactam administrées toutes les 6 heures sont résumés dans les Tableaux 5 et 6, respectivement. Les paramètres pharmacocinétiques étaient similaires pour l'administration à dose unique et répétée en raison d'une faible accumulation.
La Cmax et l'ASC de l'imipénème, la cilastatine et le relebactam augmentent proportionnellement à la dose. Les demi-vies d'élimination (t½) de l'imipénème, la cilastatine et le relebactam sont indépendantes de la dose.
Tableau 4 : Moyennes géométriques (coefficient de variation géométrique en %) des paramètres pharmacocinétiques plasmatiques à l'état d'équilibre pour l'imipénème, la cilastatine et le relebactam après plusieurs perfusions intraveineuses de 30 minutes de 500 mg d'imipénème/500 mg de cilastatine + 250 mg de relebactam toutes les 6 heures chez des adultes sains
| | Imipénème (n = 6) | Cilastatine (n = 6) | Relebactam (n = 6) |
| ASC0-6 h (µM-h) | 138,0 (17,8) | 98,0 (17,0) | 81,6 (17,8) |
| Cmax (µM) | 106,0 (26,8) | 96,4 (21,8) | 48,3 (24,9) |
| Cl (L/h) | 12,0 (17,8) | 14,2 (17,0) | 8,8 (17,8) |
| t1/2 (h)* | 1,1 (± 0,1) | 1,0 (± 0,1) | 1,7 (± 0,2) |
| *Moyenne arithmétique (écart type) rapportée pour t 1/2 ASC0-6 h = aire sous la courbe de la concentration en fonction du temps de 0 à 6 heures ; Cmax = concentration maximale ; Cl = clairance plasmatique ; t1/2= demi-vie d'élimination | |||
Tableau 5 : Modèle pharmacocinétique de population sur la base de la moyenne géométrique (coefficient de variation géométrique en %) des paramètres pharmacocinétiques plasmatiques à l'état d'équilibre pour l'imipénème et le relebactam après plusieurs perfusions intraveineuses de 30 minutes de Recarbrio (500 mg d'imipénème/500 mg de cilastatine/250 mg de relebactam) toutes les 6 heures chez des patients présentant une IIAc ou une IUc avec une ClCr égale ou supérieure à 90 mL/min
| | Imipénème | Relebactam |
| ASC0-24 h (µM-h) | 500,0 (56,3) | 390,5 (44,5) |
| Cmax (µM) | 88,9 (62,1) | 58,5 (44,9) |
| Cl (L/h) | 13,4 (56,3) | 7,4 (44,5) |
| t1/2 (h)* | 1,0 (± 0,5) | 1,2 (± 0,7) |
| *Moyenne arithmétique (écart type) rapportée pour t 1/2 ASC0-24 h = aire sous la courbe de la concentration en fonction du temps de 0 à 24 heures ; Cmax = concentration maximale ; Cl = clairance plasmatique ; t1/2= demi-vie d'élimination | ||
Tableau 6 : Modèle pharmacocinétique de population sur la base de la moyenne géométrique (coefficient de variation géométrique en %) des paramètres pharmacocinétiques plasmatiques à l'état d'équilibre pour l'imipénème et le relebactam après plusieurs perfusions intraveineuses de 30 minutes de Recarbrio (500 mg d'imipénème/500 mg de cilastatine + 250 mg de relebactam) toutes les 6 heures chez des patients présentant une PN ou une PAVM avec une ClCr égale ou supérieure à 90 mL/min
| | Imipénème | Relebactam |
| ASC0-24 h (µM-h) | 812,2 (59,4) | 655,2 (47,9) |
| Cmax (µM) | 159,1 (62,3) | 87,6 (43,8) |
| Cl (L/h) | 8,2 (59,4) | 4,4 (47,9) |
| ASC0-24 h = aire sous la courbe de la concentration en fonction du temps de 0 à 24 heures ; Cmax = concentration maximale ; Cl = clairance plasmatique | ||
Distribution
La liaison de l'imipénème et de la cilastatine aux protéines plasmatiques humaines est d'environ 20 % et 40 % respectivement. La liaison du relebactam aux protéines plasmatiques humaines est d'environ 22 % et est indépendante de la concentration.
Le volume de distribution à l'état d'équilibre de l'imipénème, la cilastatine et le relebactam est respectivement de 24,3 L, 13,8 L et 19,0 L chez des sujets après plusieurs administrations par perfusion de 30 minutes toutes les 6 heures.
La pénétration dans le liquide du revêtement épithélial (LRE) pulmonaire exprimé sous la forme du rapport du liquide de revêtement épithélial/l'exposition plasmatique libre était de 55 % et 54 % pour l'imipénème et le relebactam respectivement.
Biotransformation
Lorsqu'il est administré seul, l'imipénème est métabolisé dans les reins par la déhydropeptidase-I, donnant lieu à de faibles taux d'imipénème (en moyenne 15 à 20 % de la dose) retrouvés dans les urines chez l'Homme. La cilastatine, un inhibiteur de cette enzyme, inhibe efficacement le métabolisme rénal ; de sorte que lorsque l'imipénème est administré concomitamment à la cilastatine, des concentrations adéquates d'imipénème (environ 70 % de la dose) sont atteintes dans les urines pour permettre une activité antibactérienne.
La cilastatine est principalement excrétée dans les urines sous forme inchangée (environ 70 à 80 % de la dose) et 10 % de la dose sont retrouvés sous forme d'un métabolite N-acétyl, doté d'une activité inhibitrice contre la déhydropeptidase-I comparable à celle de la molécule mère.
Le relebactam est principalement éliminé par excrétion rénale sous forme inchangée (plus de 90 % de la dose) et est peu métabolisé. Le relebactam sous forme inchangée est la seule composante du médicament détectée dans le plasma humain.
Élimination
L'imipénème, la cilastatine et le relebactam sont principalement excrétés par les reins.
Après administration de doses répétées de 500 mg d'imipénème, 500 mg de cilastatine et 250 mg de relebactam à des hommes sains, environ 63 % de la dose d'imipénème administrés et 77 % de la dose de cilastatine administrés sont retrouvés sous forme inchangée dans les urines. L'excrétion rénale de l'imipénème et de la cilastatine implique à la fois la filtration glomérulaire et la sécrétion tubulaire active. Plus de 90 % de la dose de relebactam administrée est excrétée sous forme inchangée dans les urines chez l'Homme. La clairance rénale moyenne du relebactam est de 135 mL/min, proche de la clairance plasmatique (148 mL/min), ce qui indique une élimination pratiquement complète du relebactam par voie rénale. La clairance rénale du relebactam sous forme libre est supérieure au taux de filtration glomérulaire, ce qui suggère que, outre la filtration glomérulaire, la sécrétion tubulaire active participe à l'élimination rénale, représentant ~ 30 % de la clairance totale.
Linéarité/non-linéarité
La pharmacocinétique du relebactam est linéaire sur l'intervalle de doses allant de 25 mg à 1150 mg étudié avec une administration intraveineuse unique, et sur l'intervalle de doses allant de 50 mg à 625 mg étudié avec une administration intraveineuse de plusieurs doses toutes les 6 heures pendant 7 jours au maximum. Une accumulation minimale d'imipénème, de cilastatine ou de relebactam a été observée après plusieurs perfusions intraveineuses de 30 minutes de relebactam (50 à 625 mg) administrés de façon concomitante avec 500 mg d'imipénème/500 mg de cilastatine toutes les 6 heures pendant 7 jours au maximum chez des hommes adultes sains ayant une fonction rénale normale.
Enzymes métabolisant le médicament
Aucune étude n'a été menée pour évaluer le potentiel d'interaction de l'imipénème ou de la cilastatine avec les enzymes du CYP450.
A des concentrations pertinentes en clinique, le relebactam n'inhibe pas les CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4 in vitro dans des microsomes hépatiques humains. Le relebactam n'a présenté aucun potentiel d'induction in vitro des CYP1A2, CYP2B6 et CYP3A4 dans des hépatocytes humains. Le relebactam est donc peu susceptible de causer des interactions médicamenteuses cliniques par le biais de voies médiées par le CYP.
L'imipénème, la cilastatine et le relebactam sont tous principalement éliminés par excrétion rénale sous forme inchangée, la biotransformation constituant une voie d'élimination mineure. Recarbrio est
donc peu susceptible d'être sujet à des interactions médicamenteuses en
cas d'administration concomitante avec des inhibiteurs ou des
inducteurs du CYP.
Transporteurs membranaires
Le relebactam n'inhibe pas les transporteurs hépatiques et rénaux suivants in vitro à des concentrations cliniquement pertinentes : OATP1B1, OATP1B3, OAT1, OAT3, OCT2, P-gp, BCRP, MATE1, MATE2K, ou BSEP.
Le relebactam est activement excrété dans les urines. Il n'est pas un substrat des transporteurs OAT1, OCT2,
P-gp, BCRP, MRP2 ou MRP4, mais en est un pour les transporteurs OAT3,
OAT4, MATE1 et MATE2K. La sécrétion tubulaire active ne représentant qu'environ 30 % de la clairance totale du relebactam, il est attendu par conséquent, que l'ampleur de l'interaction médicamenteuse liée à l'inhibition des transporteurs tubulaires n'ait qu'un impact
clinique minimal, ce qui a été confirmé lors d'une étude clinique
d'interactions médicamenteuses avec le probénécide et Recarbrio (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Populations particulières
Insuffisance rénale
Dans une étude de pharmacocinétique clinique et une analyse de pharmacocinétique de population, des différences d'exposition (ASC) cliniquement significatives ont été observées pour l'imipénème, la cilastatine et le relebactam en fonction du degré d'insuffisance rénale.
Dans l'étude clinique, les moyennes géométriques de l'ASC de l'imipénème étaient jusqu'à 1,4 fois, 1,5 fois et 2,5 fois plus élevées chez les patients présentant respectivement une insuffisance rénale légère, modérée et sévère, par rapport aux sujets sains ayant une fonction rénale normale. Les moyennes géométriques respectives de l'ASC de la cilastatine étaient jusqu'à 1,6 fois, 1,9 fois et 5,6 fois plus élevées. Les moyennes géométriques de l'ASC du relebactam étaient jusqu'à 1,6 fois, 2,2 fois et 4,9 fois plus élevées chez les patients présentant respectivement une insuffisance rénale légère, modérée et sévère, par rapport aux sujets sains ayant une fonction rénale normale. Chez les patients présentant une insuffisance rénale terminale (IRT) sous hémodialyse, l'imipénème, la cilastatine et le relebactam sont efficacement éliminés par l'hémodialyse.
Afin de maintenir des expositions systémiques similaires à celles obtenues chez les sujets ayant une fonction rénale normale, une adaptation posologique est recommandée chez les patients présentant une insuffisance rénale. Les patients présentant une IRT sous hémodialyse doivent recevoir Recarbrio après la séance d'hémodialyse (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
L'imipénème, la cilastatine et le relebactam sont principalement éliminés par voie rénale ; par conséquent, l'insuffisance hépatique ne devrait pas avoir d'effet sur l'exposition à Recarbrio (voir rubrique Posologie et mode d'administration).
Personnes âgées/sexe
Dans une étude des effets en fonction de l'âge et du sexe et une analyse de pharmacocinétique de population, aucune différence d'exposition (ASC) cliniquement significative n'a été observée pour l'imipénème, la cilastatine et le relebactam en fonction de l'âge ou du sexe, en dehors de l'effet de la fonction rénale (voir rubrique Posologie et mode d'administration).
Origine ethnique
Seul un nombre limité de patients non blancs ont été inclus dans les études cliniques, mais aucun effet majeur de l'origine ethnique sur la pharmacocinétique de l'imipénème, de la cilastatine et du relebactam n'est attendu.
Recarbrio a une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. Des effets indésirables sur le SNC, tels que des convulsions, des états confusionnels et une activité myoclonique, ont été rapportés lors du traitement par imipénème/cilastatine, des composants de Recarbrio, en particulier en cas d'administration d'imipénème à des doses supérieures aux doses recommandées (voir rubrique Mises en garde et précautions d'emploi). Par conséquent, la prudence s'impose en cas de conduite de véhicules ou d'utilisation de machines.
Imipénème/cilastatine
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicologie en administration répétée et de génotoxicité n'ont pas révélé de risque particulier pour l'Homme.
Les études chez l'animal ont montré que la toxicité induite par l'imipénème, sous forme de molécule unique, était limitée au rein. La co-administration de cilastatine avec l'imipénème selon un rapport 1 : 1 a prévenu les effets néphrotoxiques de l'imipénème chez les lapins et les singes. Les données disponibles suggèrent que la cilastatine prévient la néphrotoxicité en empêchant l'entrée de l'imipénème dans les cellules tubulaires.
Dans une étude de tératogenèse chez la femelle singe cynomolgus gravide, l'administration d'imipénème/cilastatine sodique à la dose de 40/40 mg/kg/jour (injection intraveineuse en bolus) a entraîné une toxicité maternelle, incluant vomissements, manque d'appétit, perte de poids, diarrhée, avortements spontanés et mort dans certains cas. Lorsque des doses d'imipénème/cilastatine sodique (approximativement de 100/100 mg/kg/jour, soit environ 3 fois la dose intraveineuse quotidienne recommandée en clinique) ont été administrées à des femelles singes cynomolgus gravides, à une vitesse de perfusion intraveineuse simulant l'utilisation clinique chez l'Homme, l'intolérance maternelle a été minimale (vomissements occasionnels), sans mortalité maternelle ni signes de tératogénicité, mais il a été observé une augmentation de la perte embryonnaire par rapport aux groupes témoins (voir rubrique Grossesse et allaitement).
Il n'a pas été mené d'études à long terme chez l'animal pour évaluer le potentiel carcinogène de l'imipénème/cilastatine.
Relebactam
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de toxicologie en administration répétée, de toxicité sur la reproduction ou de génotoxicité n'ont pas révélé de risque particulier pour l'Homme. Aucune étude de cancérogénèse n'a été conduite avec le relebactam.
Le relebactam administré par voie intraveineuse à des rates allaitantes à une dose de 450 mg/kg/jour (JG (Jour de Gestation) 6 à JL (Jour de Lactation) 14), était excrété dans le lait à une concentration d'environ 5 % des concentrations plasmatiques maternelles.
Les études chez l'animal montrent que le relebactam administré seul a causé une dégénérescence des tubules rénaux chez des singes à une exposition plasmatique 7 fois supérieure à celle de l'exposition plasmatique chez l'Homme à la dose maximale recommandée (DMRH). La dégénérescence des tubules rénaux s'est montrée réversible après arrêt du traitement. Il n'y avait aucun signe de néphrotoxicité à des expositions plasmatiques inférieures ou égales à 3 fois l'exposition plasmatique chez l'Homme à la DMRH.
Recarbrio se présente sous forme de poudre sèche en flacon à dose unique devant être reconstituée puis diluée en utilisant une technique aseptique avant perfusion intraveineuse, comme décrit
ci-dessous :
· Afin de préparer la solution pour perfusion, le contenu du flacon doit être transféré dans 100 mL d'une solution pour perfusion appropriée (voir rubriques Incompatibilités et Durée de conservation) : chlorure de sodium à 9 mg/mL (0,9 %). Dans des cas exceptionnels où le chlorure de sodium à 9 mg/mL (0,9 %) ne peut pas être utilisé pour des raisons cliniques, le glucose à 5 % peut être utilisé en remplacement.
· Prélever 20 mL (2 x 10 mL) de solvant dans une poche pour perfusion appropriée et en introduire 10 mL dans le flacon. La suspension reconstituée ne doit pas être administrée directement par perfusion intraveineuse.
· Après reconstitution, bien agiter le flacon et transférer la suspension obtenue dans les 80 mL restant dans la poche de perfusion.
· Ajouter les 10 mL de solvant pour perfusion restants dans le flacon et bien agiter afin de s'assurer que le contenu du flacon soit complètement transféré ; répéter le transfert de la suspension obtenue dans la solution pour perfusion avant l'administration. Agiter le mélange obtenu jusqu'à ce qu'il soit limpide.
· Les solutions reconstituées de Recarbrio peuvent être incolores à jaunes. Les variations de couleur dans cette gamme n'affectent pas l'activité du produit.
· Chez les patients présentant une insuffisance rénale, une dose réduite de Recarbrio sera administrée en fonction de la ClCr du patient, comme indiqué dans le Tableau 7.
Préparer 100 mL de solution pour perfusion comme indiqué ci-dessus. Sélectionner le volume (mL) de solution pour perfusion finale nécessaire pour la dose appropriée de Recarbrio comme indiqué dans le Tableau 7.
Les médicaments pour administration parentérale doivent être contrôlés visuellement afin de vérifier l'absence de particules et de coloration anormale avant l'administration, dès lors que la solution et son contenant le permettent. Jeter la solution en cas de coloration anormale ou de particules visibles.
Tableau 7 : Préparation des doses de Recarbrio
|
Clairance de la créatine (mL/min) |
Posologie (mg) de Recarbrio (imipénème/cilastatine/relebactam) |
Volume (mL) de solution à prélever et à éliminer de la préparation |
Volume (mL) de solution pour perfusion finale nécessaire pour l'administration |
|
Supérieure ou égale à 90 |
500/500/250 |
N/A |
100 |
|
Inférieure à 90 et supérieure ou égale à 60 |
400/400/200 |
20 |
80 |
|
Inférieure à 60 et supérieure ou égale à 30 |
300/300/150 |
40 |
60 |
|
Inférieure à 30 et supérieure ou égale à 15 ou IRT sous hémodialyse |
200/200/100 |
60 |
40 |
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicaments compatibles
La compatibilité physique de Recarbrio avec certains médicaments injectables a été évaluée dans deux solvants d'utilisation courante au niveau d'un site de perfusion en Y. Les médicaments compatibles avec le solvant compatible correspondant (c'est-à-dire solution injectable de dextrose à 5 % ou de chlorure de sodium à 0,9 %) sont listés ci-dessous. Recarbrio ne doit pas être administré de façon concomitante au moyen de la même ligne intraveineuse (ou canule) que d'autres médicaments non listés ci-dessous car aucune donnée sur la compatibilité n'est disponible. Consulter respectivement les informations sur l'utilisation du (ou des) médicament(s) administré(s) de façon concomitante pour vérifier la compatibilité en vue d'une administration simultanée. Ce médicament ne doit pas être mélangé à d'autres médicaments à l'exception de ceux mentionnés ci-dessous.
Liste des médicaments injectables compatibles pour utilisation avec un solvant injectable de dextrose à 5 % ou de chlorure de sodium à 0,9 %
· dexmédétomidine
· dopamine
· adrénaline
· fentanyl
· héparine
· midazolam
· noradrénaline
· phényléphrine
Matériaux de poches et kits pour perfusion intraveineuse compatibles
Recarbrio est compatible avec les matériaux des poches et kits pour perfusion intraveineuse suivants. Tout matériau de poche ou kit pour perfusion intraveineuse non listé ci-dessous ne doit pas être utilisé.
Matériaux de poches pour perfusion intraveineuse
Polychlorure de vinyl (PVC) et polyoléfine (polypropylène et polyéthylène)
Matériaux de kits pour perfusion intraveineuse (avec tubulure)
PVC
+ Di-(2-éthylhéxyl)phtalate (DEHP) et PVC avec un
revêtement en polyéthylène (PE)
Médicaments incompatibles
Recarbrio pour solution pour perfusion est physiquement incompatible avec le propofol dans du dextrose (également appelé glucose) à 5 % ou du chlorure de sodium à 0,9 %.
Liste I
Prescription hospitalière.
Poudre pour solution pour perfusion.
Poudre de couleur blanche à jaune pâle.
Flacon en verre de 20 mL, fermé par un bouchon en caoutchouc de 20 mm et serti avec une capsule en aluminium.
Le médicament est présenté en boîtes de 25 flacons.
Chaque flacon contient de l'imipénème monohydraté équivalent à 500 mg d'imipénème, de la cilastatine sodique équivalent à 500 mg de cilastatine, et du relebactam monohydraté équivalent à 250 mg de relebactam.
Excipient(s) à effet notoire
La quantité totale de sodium dans chaque flacon est de 37,5 mg (1,6 mmol).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Hydrogénocarbonate de sodium